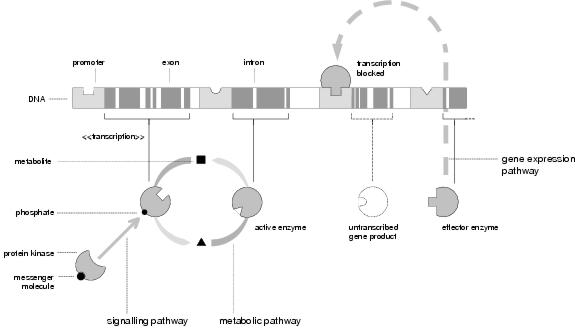
Figure 4.1: Biochemical Pathways
Having introduced the key components of biological systems - cells, genes and proteins - these elements can now be bought together to describe higher-level structures which form the basis of control and function within biological organisms. These structures, called biochemical pathways, form networks of computation and communication which permeate organisms, bringing about order through the control and co-ordination of the activity of local systems.
Figure 4.1: Biochemical Pathways
There are three categories of biochemical pathway (see figure 4.1). Metabolic networks exist within cells, emerging from interactions between locally-transcribed proteins. Signalling networks comprise cellular responses to inter-cellular signals. Gene expression networks describe regulatory interactions between genes and gene-products. Marijuán [1995] describes these activities, respectively, as self-organisation, self-reshaping and self-modification. Self-organisation reflects how a distributed system of independent components can carry out a unified process. Self-reshaping is the ability of an existing system to carry out multiple processes to satisfy varying needs. Self-modification is the ability of a system to change its constitution in order to solve unforeseen problems. Interactions between the three classes of pathway unifies these processes and bring out the emergent behaviour of the whole organism.
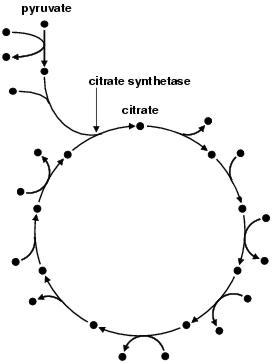
Figure 4.2: The citrate cycle. Nodes represent products and substrates. Arrows represent enzymes.
The previous chapter introduced the structure and function of individual proteins and enzymes, their capacity to interact - through sharing of products and substrates - and their mechanisms of regulation: activation, inhibition and allosterism. This section discusses the intra-cellular structures which emerge as a result of these interactions.
Figure 4.2 illustrates one such structure, the citrate cycle12, a central part of the metabolism of both eukaryotes and prokaryotes (although not always used in the same way, see Forst and Schulten, [1999]). The citrate cycle, in turn, fits into a larger structure, the respiratory chain, a sequence of reactions which eventually converts food into energy. Feeding the citrate cycle are other pathways, most notably glycolysis; which converts carbohydrates into pyruvate. Pyruvate is then converted into NADH by the citrate cycle, which feeds later stages of respiration. Pathways are highly evolved, conservative, structures which often carry out more than one task at once. For example, in addition to its role in respiration, the citrate cycle produces intermediates used in amino acid synthesis.
A metabolic pathway consists of two interdependent flows: reaction pathways and control feedbacks. Reaction pathways are composed of systems of enzymes with tightly linked specificities for one another's products and substrates. In many cases, these pathways are forked, with more than one enzyme having specificity for a given product. As demonstrated by the citrate cycle, pathways may also feed back into themselves, producing an iterative structure. Control feedback may be positive or negative, internal to the pathway, or caused by external metabolic or signalling pathways. The citrate cycle is regulated by substrates and products within the cycle. Glycolysis is regulated by hormones, notably insulin. The functions of pathways are either anabolic (constructive), catabolic (destructive), or amphibolic. Amphibolic pathways are both anabolic and catabolic, and often link anabolic and catabolic pathways.
The sharing of reaction and control between pathways means that most pathways are not separate structures, but conventions which indicate one way of subdividing the metabolism into smaller systems. However, some pathways (or parts of pathways) are independent of others. These are genetically independent pathways [Schilling et al., 1999]: pathways where there exist no other pathways which use only a subset of their reactions13. Approximately 80% of metabolic intermediates have just one use in the cell [Marijuán, 1994]. However, the existence of regulatory networks makes it unlikely that genetically independent pathways will be long, since at some point a pathway is regulated by factors in another pathway. To make understanding of the pathway easier, convention defines pathways such that they largely contain a regulatory network.
Even though many key reaction pathways have been identified and charted, the metabolism is still not completely understood. As a result, known pathways tend to reflect incomplete knowledge, describing a subset of the operation of actual metabolisms. Factors such as re-use of reactions, multiple equivalent paths through the network, and variety between cell types and between individuals (since our genomes are not identical) add to the difficulty of understanding. Fortunately, with the genomes of a number of organisms now completely sequenced, the data required to fuel this understanding is becoming available. The difficult task now is in interpreting this data, a task undertaken by traditional mathematical techniques [Heinrich and Schuster, 1998], modelling [Reddy et al., 1993,Reddy et al., 1996], and bioinformatics [Schilling et al., 1999]. A better understanding of the metabolism would have many uses, including those in bio-process engineering (creation of novel metabolic pathways for use in chemical plants) and health-related (finding alternative paths through defective networks, design of drugs which intervene in the metabolism) areas [Karp and Mavrovouniotis, 1994]. Current understanding is expressed through charts and diagrams [Michal, 1999] (for which representation is a key concern [Michal, 1998,Karp and Paley, 1994]) and public databases [Karp, 1999,European Bioinformatics Institute, 2000].
Finally, understanding of the metabolism can be aided by an understanding of how it evolved. This topic has been approached from a number of areas, including biochemistry [Lyubarev and Kurganov, 1997,Wächtershäuser, 1990], biophysics [Igamberdiev, 1999] and molecular evolution [Page and Holmes, 1998,Forst and Schulten, 1999]. The molecular evolution approach uses the current form of genomes to infer properties of earlier forms. For example, in many cases enzymes proximal in a pathway are also proximal in the genome, since they must have had a high degree of genetic linkage in order to have survived recombination events during evolution.
Signalling between cells in an organism is a fairly well understood process. Cells produce signal proteins which, after being released through the cell's plasma membrane, spread throughout the organism via bodily fluids. Receptors on the surface of other cells then detect this signal and relay it to the appropriate components within the cell. How this intra-cellular communication happens, by comparison, is only now beginning to be understood by molecular biologists.
In fact, research shows this process to be surprisingly complex - not simply a case of a receptor generating the right signal and transmitting this into the cytoplasm - but rather an extended series of events involving many different agents. Key amongst these agents are protein kinases and phosphotases, enzymes which modify the activity of other enzymes. Other players include secondary messenger molecules, protein adapters - which connect other proteins together - and scaffold proteins - which co-ordinate the activities of other molecules.
Under the classic model [Sutherland, 1972,Fisher et al., 1999], occupation of a receptor by an external signal molecule causes the activation of an internally-bound effector enzyme. This effector then generates secondary messenger molecules which spread the signal throughout the cell and activate target proteins. These signal sensitive protein kinases or phosphotases then, either directly or indirectly via the (in)activation of other protein kinases or phosphotases, modify proteins within the metabolic or gene expression networks of the cell. This brings about a change in cellular activity. Of particular note is the degree of amplification involved in the system: one primary signal molecule causes many secondary signal molecules which then activate a hierarchy of modifier proteins.
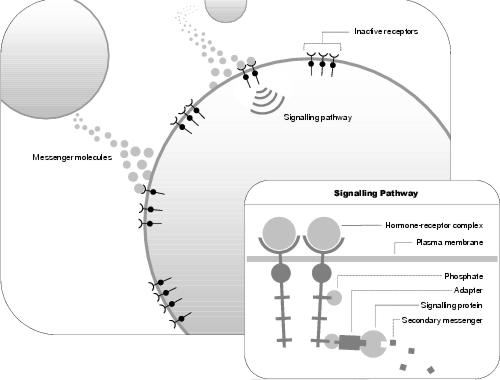
Figure 4.3: Initiation of a Signalling Pathway
More recent research (summarised in [Scott and Pawson, 2000]) refines this understanding. Many receptors are in the family receptor tyrosine kinase. These enzymatic proteins work in pairs. When a pair of receptors bind signal hormones, their cytoplasmic tails (which protrude below the plasma membrane) bind phosphate molecules to one another's tyrosine amino acids. These altered residues then become bound, either directly, or indirectly through adapters, to signalling enzymes - activating the signalling enzymes, causing them to generate secondary messenger molecules (see figure 4.3). The key differences in this new interpretation are that: (i) signalling proteins typically consist of multiple domains; and (ii) these proteins are spatially co-ordinated. In more complicated signalling systems, scaffold proteins, which consist of multiple linker domains, provide this co-ordination. Indeed, they are particularly prevalent in hierarchical protein kinase/phosphotase systems. This is necessary because these enzymes often have broad specificity, meaning that if not co-ordinated, they can modify the activity of a wide range of proteins - an effect not typically wanted. In addition, the modular make-up of signalling proteins is thought to improve the evolvability of these systems, since new systems can be formed by novel re-arrangements of existing, and newly created, modules.
Bray [Bray and Lay, 1994,Bray, 1995] has described computational aspects of signalling pathways. Enzymes involved in signalling pathways can be conceptually seen as having a number of inputs, each corresponding to a certain class of regulatory or phosphorylation events. The action (output state) of the enzyme is then some function of these inputs. For instance, enzymes exist which become active only after being phosphorylated by two protein kinases - an AND function; whereas some require the presence of only one of two possible regulatory molecules: an OR function. A more general pattern is that of amplification and switching i.e. if a signal (one molecule) activates an enzyme, the enzyme reacts with an amplified secondary signal (many molecules). Combining these behaviours - function application, switching and amplification - results in a system not unlike a neural network. In fact, signalling enzymes are more like McCulloch-Pitts neurons (perceptrons) than are real neurons. Taking this idea further, Bray has experimented with abstract models of these `protein circuits', using simulated evolution (optimising reaction rates and binding constants) as a training system.
The switching behaviour of an enzyme depends upon a number of kinetic properties: properties which affect the speed and crispness of responses. However, the quantum mechanical behaviour of enzymatic reactions means that they do not always behave as ideal Boolean switches, but display varying degrees of fuzzy response. Bray suggests that this behaviour is caused by asymmetries and synergisms between inputs; and results in enzymes behaving like fuzzy logic elements, which may be more computationally powerful than Boolean logic units. Fisher et al. [1999], too, discuss the fuzzy properties of signalling enzymes, particularly protein kinases, comparing their behaviour to fuzzy classifier systems. Moreover, they describes the `signalling ecology' as a "vast parallel distributed processing network of adaptable agents", conferring such cognitive properties as pattern recognition, handling fuzzy data, memory capacity and context-sensitivity to these agents.
A cell's type is defined by the subset of genes which are typically expressed (transcribed) within the cell. Recall from section 3.2.2 that transcription of a particular gene depends upon the correct formation and activity of a transcription complex, a process which can be blocked by the binding of certain inhibitors to regulation sites associated with the gene. Inhibitors are proteins and therefore must be encoded by other genes. Each of these regulating genes, in turn, may be regulated by inhibitors encoded by other genes. Typically, a single regulating gene may regulate a small number of other genes, each of which may then, in turn, regulate a small number of other genes. By transitivity, a single gene may indirectly affect the regulation of a large number of other genes. A gene may also, either directly or indirectly, regulate its own transcription. This complex system of regulatory interactions is known variously as a gene expression network, a genomic regulatory network, or more concisely, a genetic network.
Genetic networks are not easy to understand and, like all discrete dynamical networks, are difficult to analyse using partial differential equations and other techniques from continuous mathematics. Hence, they are typically modelled with automata and analysed by simulation, a trend captured by Stuart Kauffman in the form of random boolean networks (RBN's) [Kauffman, 1969,Somogyi and Sniegoski, 1996]. These describe regulatory networks as a mapping from state t to state t+1, employing directional links, binary on/off states, parallel updates and combinatorial logic on inputs. Whilst an idealised representation, these abstract networks still demonstrate behaviour similar to real networks. Interestingly, when simulated, the networks indicate the existence of attractors, which pull the network towards a small set of final states starting from a large number of initial states. In [Somogyi and Sniegoski, 1996,Wuensche, 1998], this behaviour is compared to specialisation in cells with an attractor comparable to a single cell type and paths between attractors equivalent to pathways of differentiation. Following from this, a cell type can be expressed as a set of closely related gene expression patterns, centered around a basin of attraction. It is also conjectured that the strength of this attractor must represent a balance between a cell's adaptiveness and a need not to cross to neighbouring states (which could, for instance, correspond to cancer cells).
As well as being dynamic and self-adaptive in their own right, networks of genetic expression are also directly subject to evolution. In [Shimeld, 1999], this evolution is discussed in terms of gene duplication events. In [Bornholdt and Sneppen, 1998,Bornholdt and Rohlf, 2000] artificial evolution is used to evolve connectivity in Boolean (and other dynamical) networks. In [Bignone et al., 1997], evolution of genetic networks is considered from a mathematical perspective.
Chiva and Tarroux, [1995], too, have applied simulated evolution in order to gain a greater understanding of how biochemical networks develop. Their system includes both transcriptional (gene regulation) and post-translational (protein interactions) components, reflecting the coupling between genes and gene-products - and the influence of both in the gene regulation process. These `protein regulation networks' are deterministic, recurrent and synchronous. The activation state (concentration) of a unit (protein) is a weighted sum of activations of other units. The weights are defined in a genome, where a gene represents each protein. Gene expression is defined by two sets: the set of states where this protein may be regulated by others, and the set of states where this protein may regulate others. The intent is that the model should mimic protein folding. The algorithm used is a genetic algorithm with uniform crossover, a degree of steady-state and a set (no duplicates) as the population structure. The last point is meant to reduce genetic drift. Gene duplication and new gene events take place population-wide at fixed intervals (keeping all solutions the same length). The system is intended to emulate differentiation, triggered by external stimuli. Hence, the fitness function rewards solutions which change state when given pertinent stimuli and punishes those which differentiate with false or vacant signals. Communication signals are implemented with `clamped' units, whose concentration (activation) is externally dictated.
A key observation is that a gradual increase in solution size from N0 to N1 produced markedly superior results than fixing solution size at either N0 or N1. This suggests that gene networks evolve best by building upon existing, smaller, networks - an observation that explains why yeast and humans share similar pathways; the human pathways including the yeast pathways as subsets of their possible reactions. The explanation behind this phenomenon is that evolution in smaller networks is much more likely to find the local optimum, whereas evolution in a larger network is harder. For a larger network to evolve, it must go via stable intermediate states - which are provided explicitly by smaller networks.
Another observation is the development of `command units', those for which fan-out is of much higher intensity than fan-in - amplification - and for whom input signals are heterogenous, whereas output signals are homogenous. This causes the formation of hierarchies, similar to those found in biological signalling pathways. Finally, the genome encoding used implicitly made the network fully-connected (although some of these connections may be ignored by a given unit). The authors speculate that sparsely-connected networks, corresponding to biological systems, might provide a better opportunity to develop clusters of specialised function within the network.
12Also known as the Krebs (after its discoverer) cycle and the tricarboxylic acid cycle
13The term genetically independent stems from the fact that the enzymes in this pathway define an independent genotype.